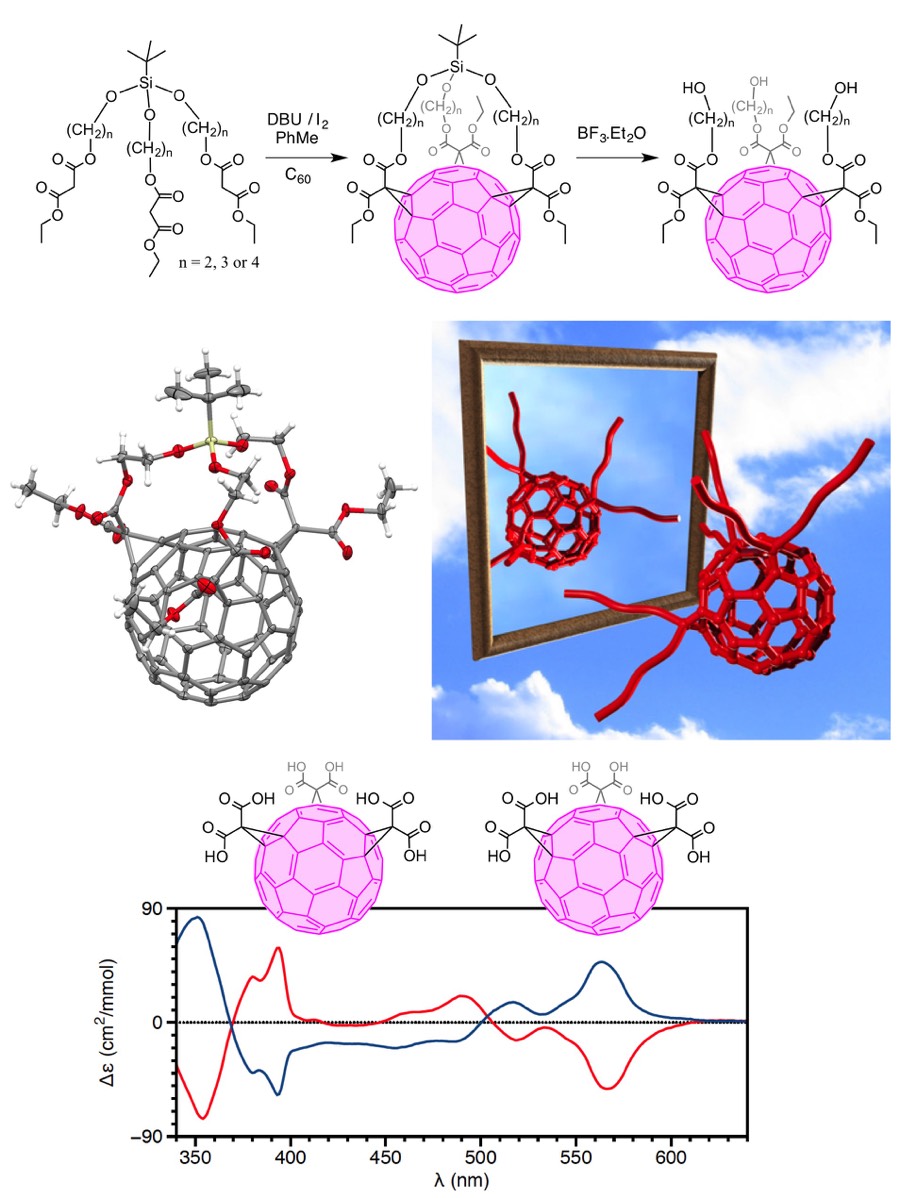
Fullerene chemistry has generated unprecedented stereochemical problems and the regioselective polyfunctionalization of [60]fullerene through multiple addition reactions remains a major challenge. Actually, mono-functionalized [60]fullerene derivatives possess nine different 6-6 bonds (bonds at the junction between two six-membered rings) and mixtures of regioisomers are obtained by successive reactions at the [60]fullerene core. The synthesis of fullerene tris-adducts is even more challenging as 46 isomers differing by the relative position of the three addends on the carbon sphere are theoretically possible. As part of this research, we have developed an expeditious synthesis of fullerene tris-adducts based on a threefold Bingel reaction between [60]fullerene and t-butyl(trialkoxy)silane derivatives bearing three malonate substituents. The silane unit is at the same time a directing group allowing the control of the stereochemistry of the tris-addition on the [60]fullerene sphere during the cyclisation step and a protecting group that can be readily cleaved to generate the corresponding acyclic fullerene derivatives. Based on this finding, we have also shown that the reaction of optically active Si-tethered tris(malonates) with [60]fullerene provides easily separable diastereoisomeric [60]fullerene tris-adducts differing by the absolute configuration of the inherently chiral e,e,e addition pattern on the fullerene core. Subsequent ester hydrolysis of the diastereoisomeric cyclic fullerene derivatives thus obtained gave optically pure tris(malonic acid) derivatives of [60]fullerene.
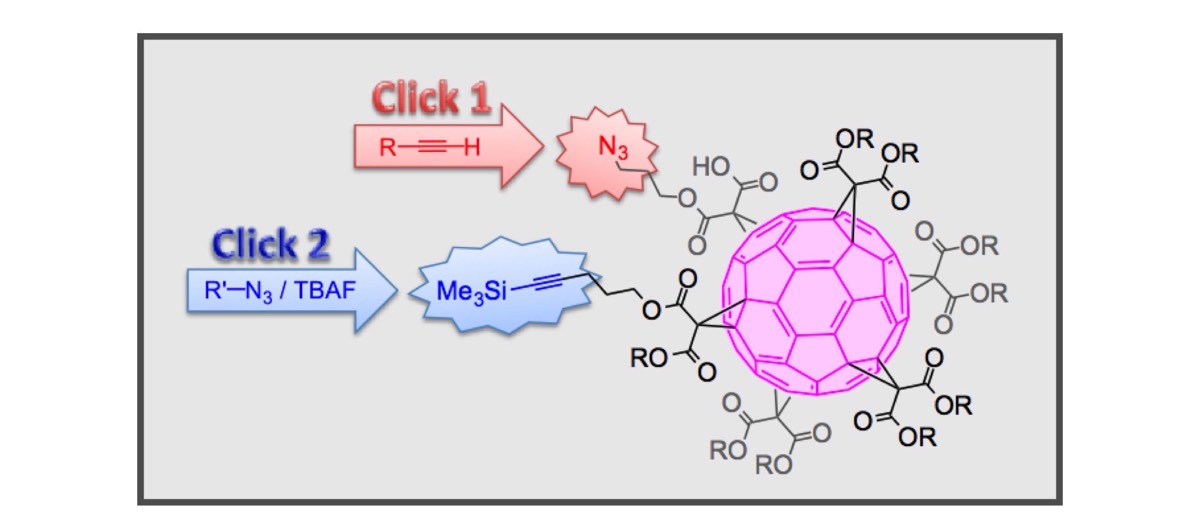
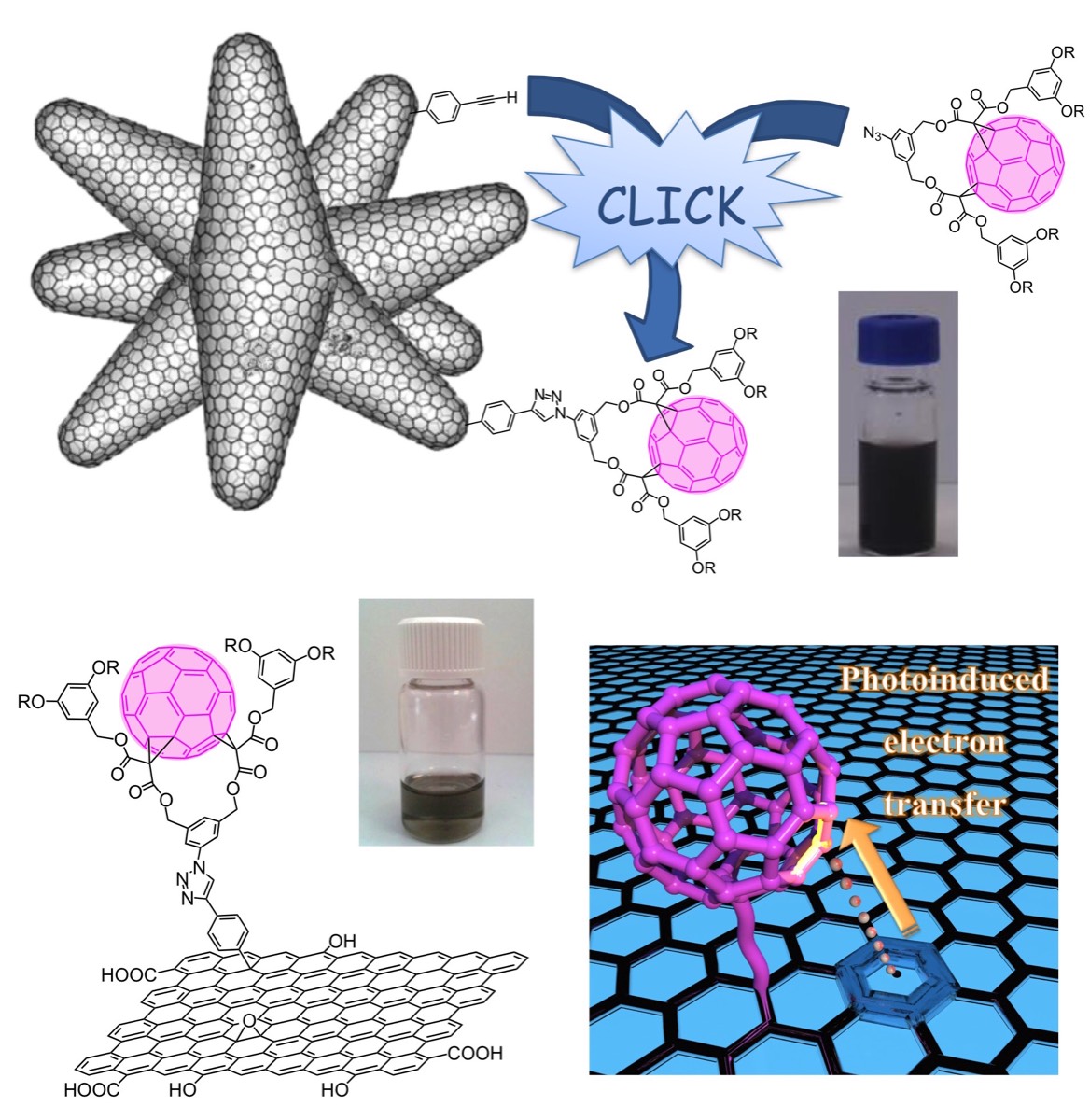
The concept of click chemistry introduced by Sharpless contributed to popularize specific synthetic tools in different branches of science. This is the case of the copper-catalyzed alkyne-azide 1,3-dipolar cycloaddition (CuAAC) reaction. This emblematic click reaction largely crossed the borders of organic chemistry and is now at the forefront of many interdisciplinary studies at the interfaces between chemistry, physics and biology. In 2008, our group became interested in evaluating the potential of the CuAAC reaction in the field of fullerene chemistry. The initial driving force for this research was not simply associated with the use of the CuAAC reaction itself for the synthesis of new fullerene derivatives. Actually, one of the most important aspects was related to the new possibilities offered by this reaction for the preparation of new compounds that were not easily accessible by using the classical tools of fullerene chemistry. This is in particular the case for fullerene hexa-adducts with a Th-symmetrical addition pattern. Indeed, such fullerene derivatives are obtained in one step from the reaction of [60]fullerene with malonates but the reaction conditions are very sensitive to steric effects and only produce the expected hexa-functionalized fullerene derivatives in reasonable yields when starting from quite simple malonates. In order to overcome this problem, various [60]fullerene hexa-adducts bearing twelve terminal groups allowing their further functionalization have been prepared. The CuAAC reaction is an extremely powerful tool for their post-functionalization.
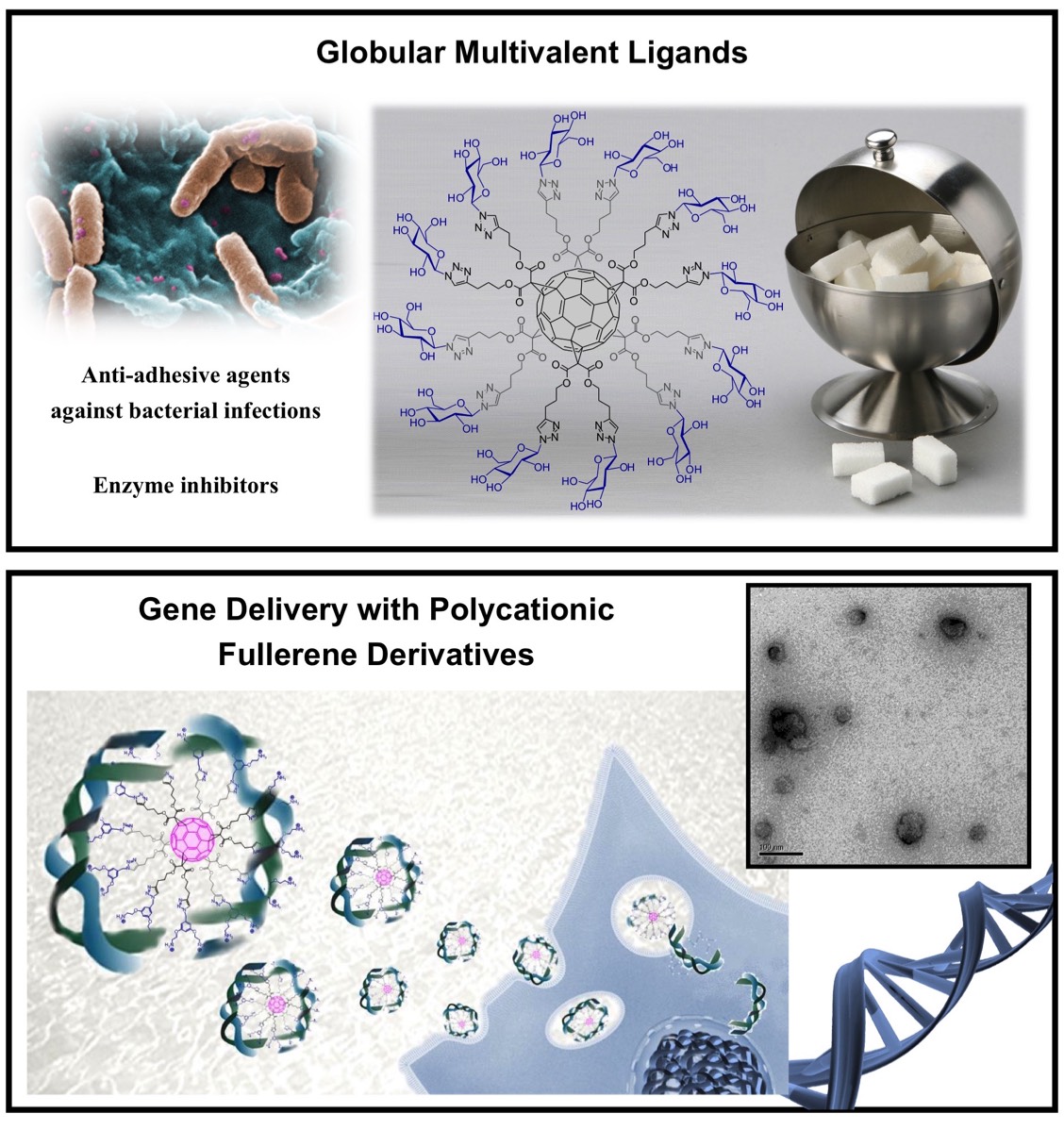
Whereas materials science applications of fullerenes have focused an enormous attention, medicinal chemistry of fullerenes has been somehow limited by the absence of solubility of most fullerene derivatives in aqueous media. The development of efficient strategies to obtain biocompatible fullerenes has been a central aspect of this field of research for many years. A large variety of water soluble fullerene derivatives have been prepared and spectacular findings reported about their biological properties. As part of this research, our clickable building blocks have been used to prepare bioactive fullerene derivatives. For example, globular glycofullerenes, i.e. fullerene sugar balls, have been constructed on the hexa-substituted fullerene scaffold. The high local concentration of carbohydrates around the carbon core in fullerene sugar balls is perfectly suited to the binding of lectins through the "glycoside cluster effect" and these compounds are potential anti-adhesive agents against bacterial infection. Moreover, when substituted with peripheral iminosugars, dramatic multivalent effects have been observed for glycosidase inhibition. These unexpected observations have been rationalized by the interplay of interactions involving the catalytic site of the enzymes and non-glycone binding sites with lectin-like abilities. Finally, polyplexes prepared from DNA and globular compact polycationic derivatives constructed around a fullerene hexakis-adduct core have shown remarkable gene delivery capabilities.
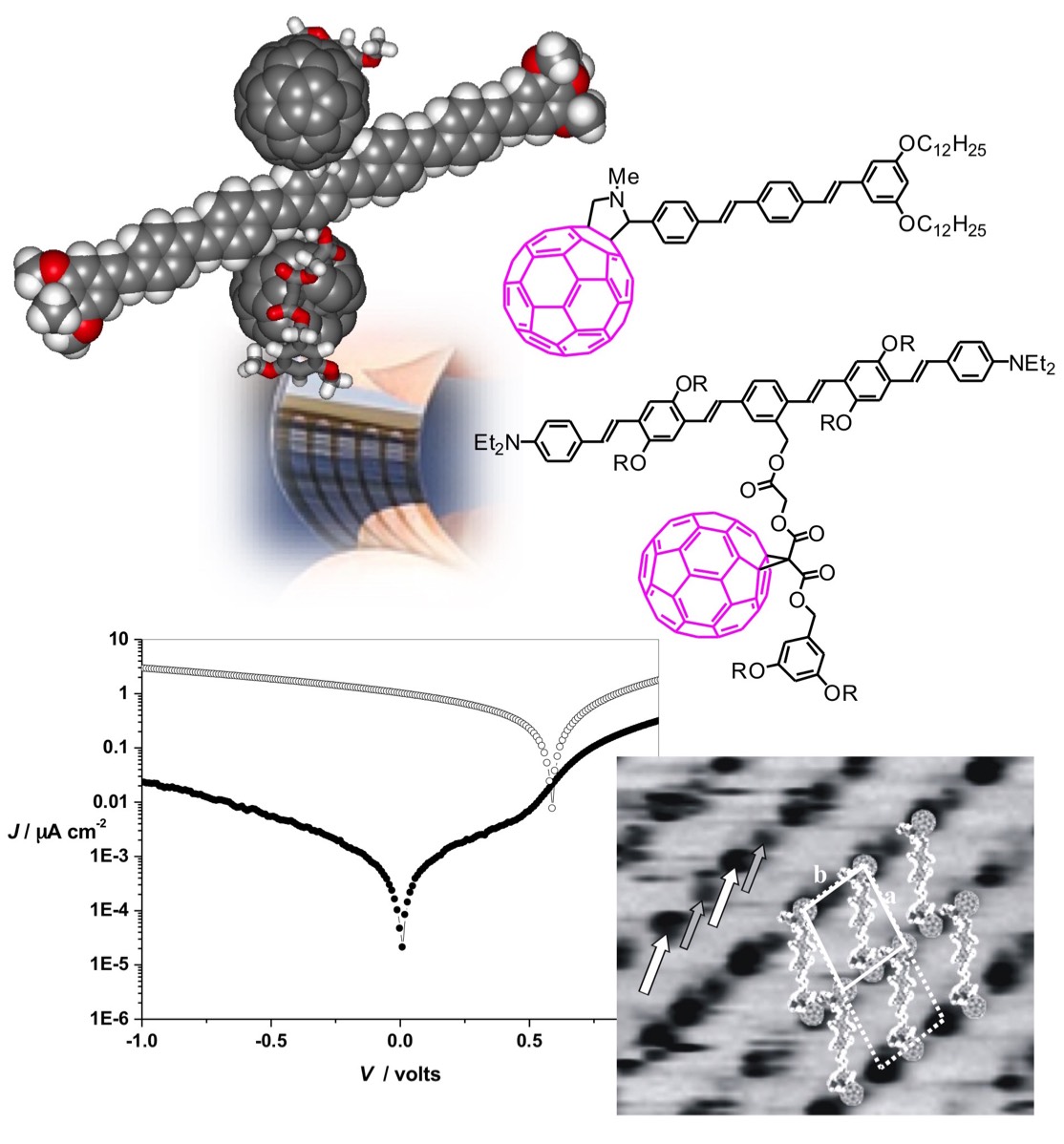
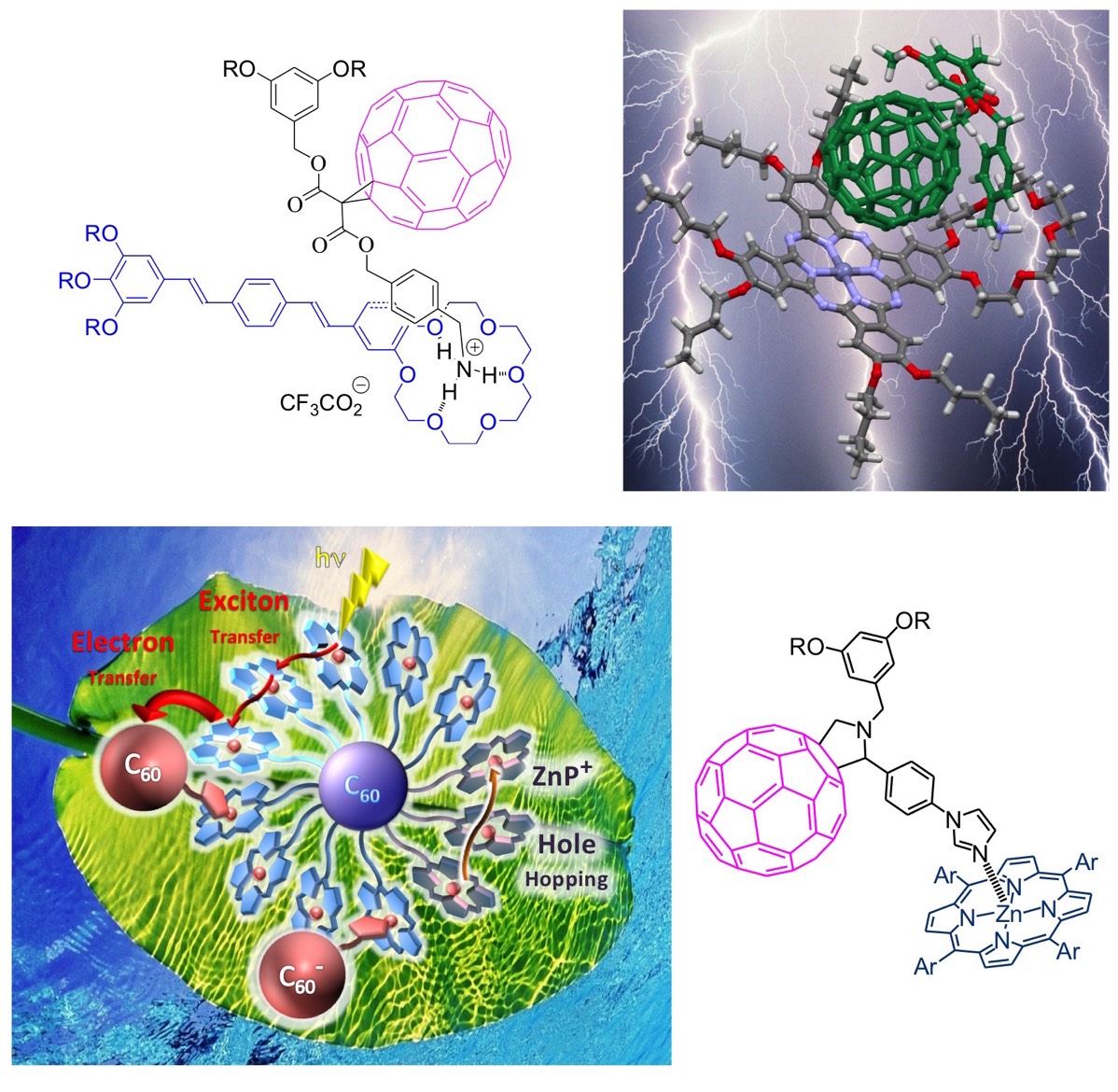
The unique electronic properties of [60]fullerene have generated significant research activities focused on its use as an electron and/or energy acceptor in photochemical molecular devices. Our group has significantly contributed to this field and various series of donor-fullerene arrays have been developed. In the particular case of covalent systems combining the carbon sphere with π-conjugated oligomers, we have been the first to show that such compounds can be incorporated into photovoltaic devices. The molecular approach for solar energy conversion is particularly interesting since the bicontinuous network obtained by chemically linking the hole-conducting moiety to the electron-conducting fullerene subunit prevents any problem arising from the effective donor/acceptor interfacial area, as observed for polymer/fullerene blends. In addition, another major advantage of our approach is that the behavior of a unique molecule in a photovoltaic cell allows easy determination of structure/activity relationships for a better understanding of the photovoltaic system. Over the years, we have contributed to elucidating the key structural parameters allowing the design of fullerene-(π-conjugated oligomer) dyads capable of achieving efficient and fast photoinduced charge separation, a critical point for the development of new molecular photovoltaic materials.
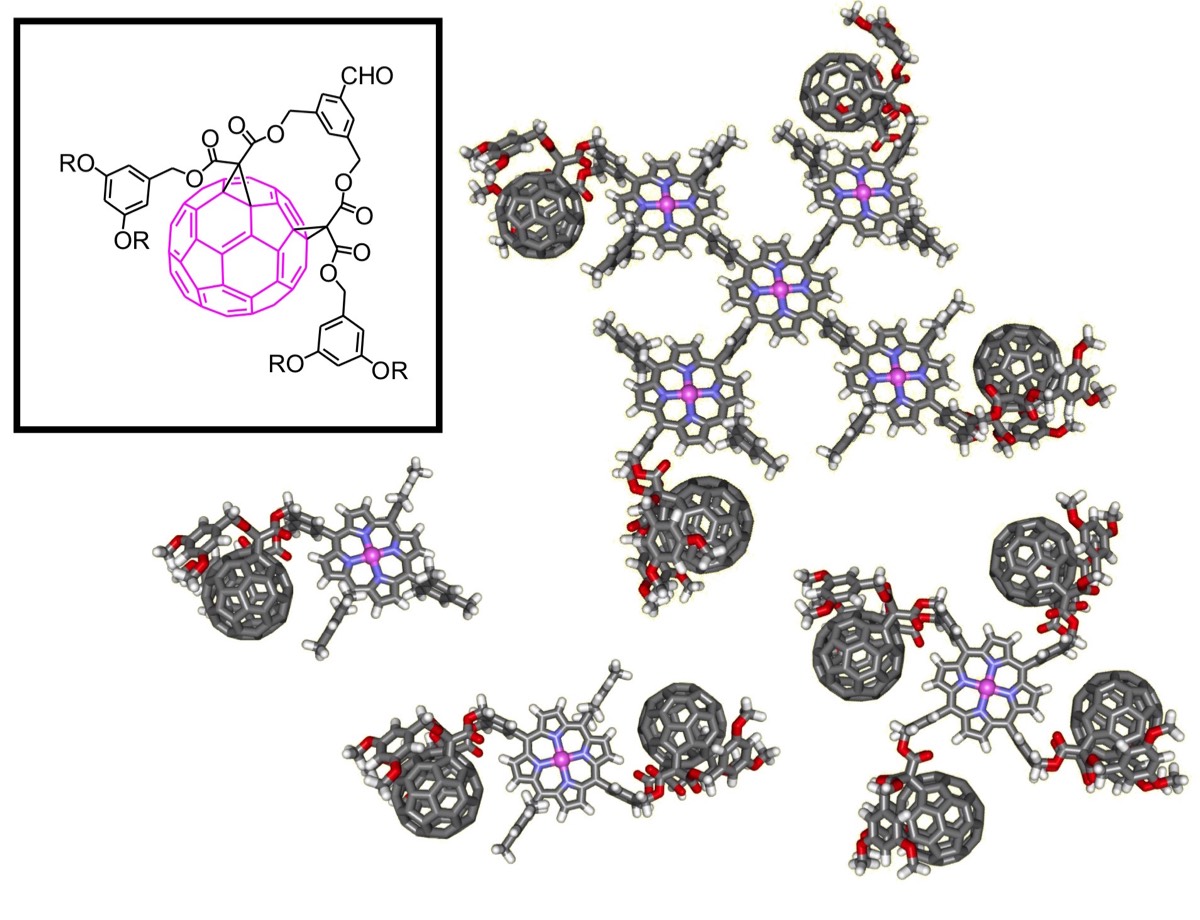
Porphyrins and fullerenes are also interesting complementary building blocks for the preparation of artificial photosynthetic systems. Indeed, many examples of covalently linked porphyrin-fullerene derivatives have been described in the literature. As far as their synthesis is concerned, porphyrin-fullerene dyads are usually obtained by reaction of a preconstructed porphyrin derivative with [60]fullerene itself or a fullerene derivative. In contrast, the use of fullerene building blocks in porphyrin synthesis has been scarcely considered. This is mainly associated with the chemical reactivity of the fullerene moiety. Effectively, fullerene derivatives react readily with radicals, various nucleophiles, carbenes, and participate as reactive 2π component in a variety of cycloaddition reactions. Thus the range of reactions that can be used for the further transformations of fullerene-containing building blocks appears to be quite limited and the compatibility of fullerene derivatives with reaction conditions classically used for porphyrin synthesis was not obvious. Indeed, we have shown that porphyrin chemistry can be performed from fullerene-benzaldehyde derivatives thus allowing us to prepare sophisticated photoactive molecular devices.
As part of this research, we became also interested in the synthesis of soluble hybrid material that combines fullerenes and other carbon nanoforms into a single covalent structure. The conjugation of fullerene with other carbon allotropes is however difficult due to the low solubility of the starting materials. Moreover, it is also limited by the chemical reactivity of the fullerene subunit and most of the reactions typically used for the functionalization of carbon nanohorns or graphene oxyde are not compatible with fullerene derivatives. Indeed, we have shown that hybrid nanostructures combining fullerenes with other carbon nanoforms can be efficiently obtained by the pre-functionalization of the carbon nanomaterial with terminal alkyne subunits followed by the grafting of a fullerene-azide under CuAAC conditions. This work was carried out in collaboration with the group of Fernando Langa (Toledo, Spain).
Whereas research focused on the use of [60]fullerene as the acceptor in covalently bound donor-acceptor pairs has received considerable attention, only a few related examples of fullerene-containing non-covalent systems have been described so far. The assembly of the two molecular components by using supramolecular interactions rather than covalent chemistry appears however particularly attractive since the range of systems that can be investigated is not severely limited by the synthetic route. As part of this research, we have developed a non-covalent approach based on the self-assembly of [60]fullerene derivatives bearing an ammonium unit with crown ethers for the preparation of photoactive supramolecular systems. On the other hand, we also use the apical coordination of metalloporphyrins with fullerene-imidazole derivatives to self-assemble sophisticated systems mimicking the natural photosynthetic system.
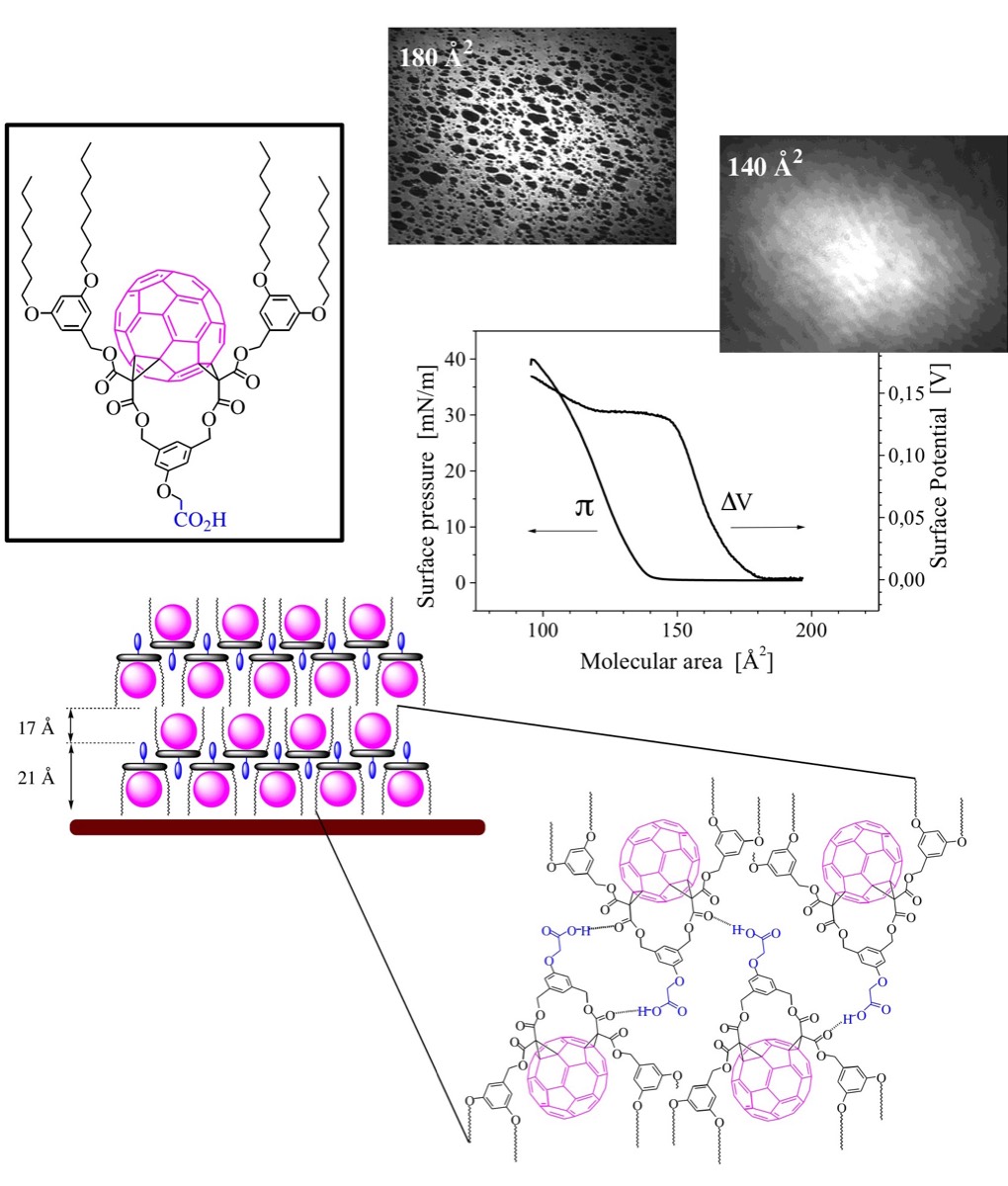
The incorporation of fullerenes into thin films is required for the preparation of many optoelectronic devices. One possible approach towards structurally ordered fullerene assemblies is the preparation of Langmuir films at the air-water interface and their subsequent transfer onto solid substrates. As part of this research, we have reported some of the fundamental architectural requirements needed for the design of amphiphilic fullerene derivatives capable of forming stable Langmuir films. Specifically, the encapsulation of the fullerene sphere in a cyclic addend surrounded by long alkyl chains is an efficient strategy to prevent the irreversible aggregation resulting from the strong fullerene-fullerene interactions usually observed for amphiphilic fullerene derivatives at the air-water interface.
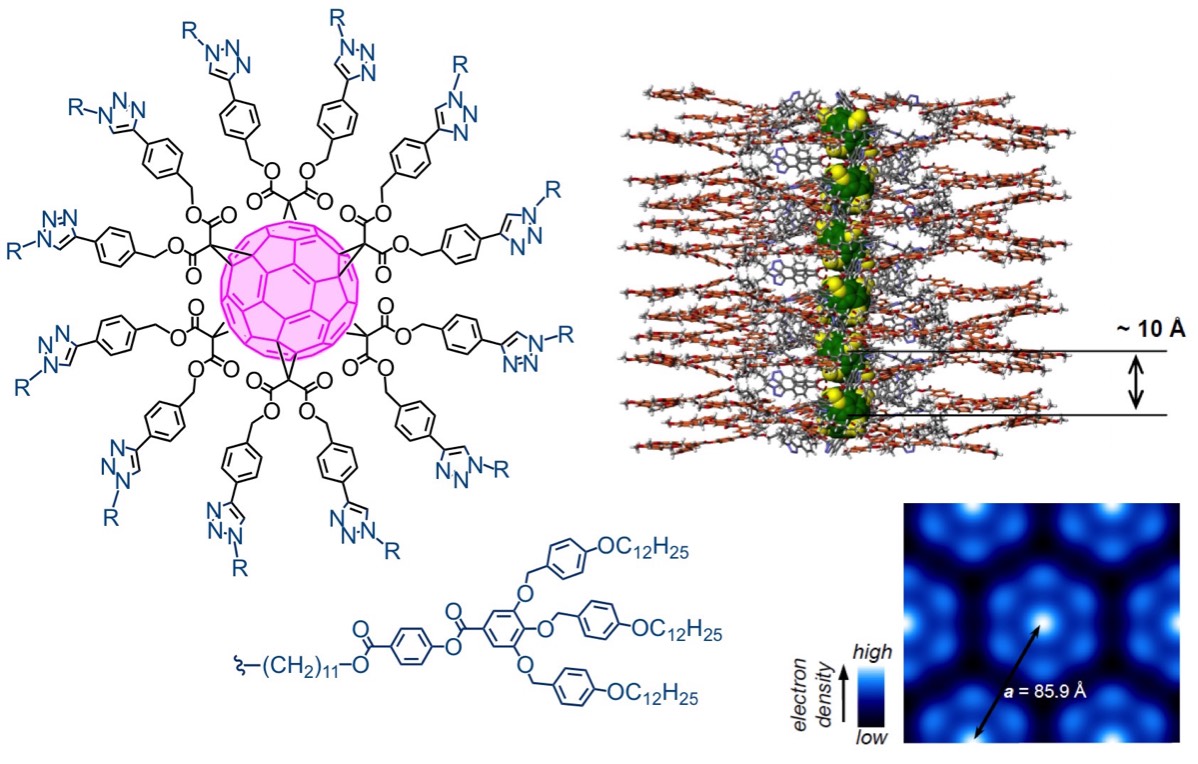
The group has also some expertise in the design of liquid-crystalline fullerene derivatives. For example, we have recently used our fullerene scaffolds to prepare dendronized derivatives. The structure adopted by these compounds was determined by the self-assembling peripheral dendrons. These twelve dendrons mediate the self-organisation of the dendronized [60]fullerene into a disc-shaped structure containing the [60]fullerene in the centre. The fullerene-containing discs self-organise into supramolecular columns with a fullerene nanowire-like core, forming a 2D columnar hexagonal periodic array.
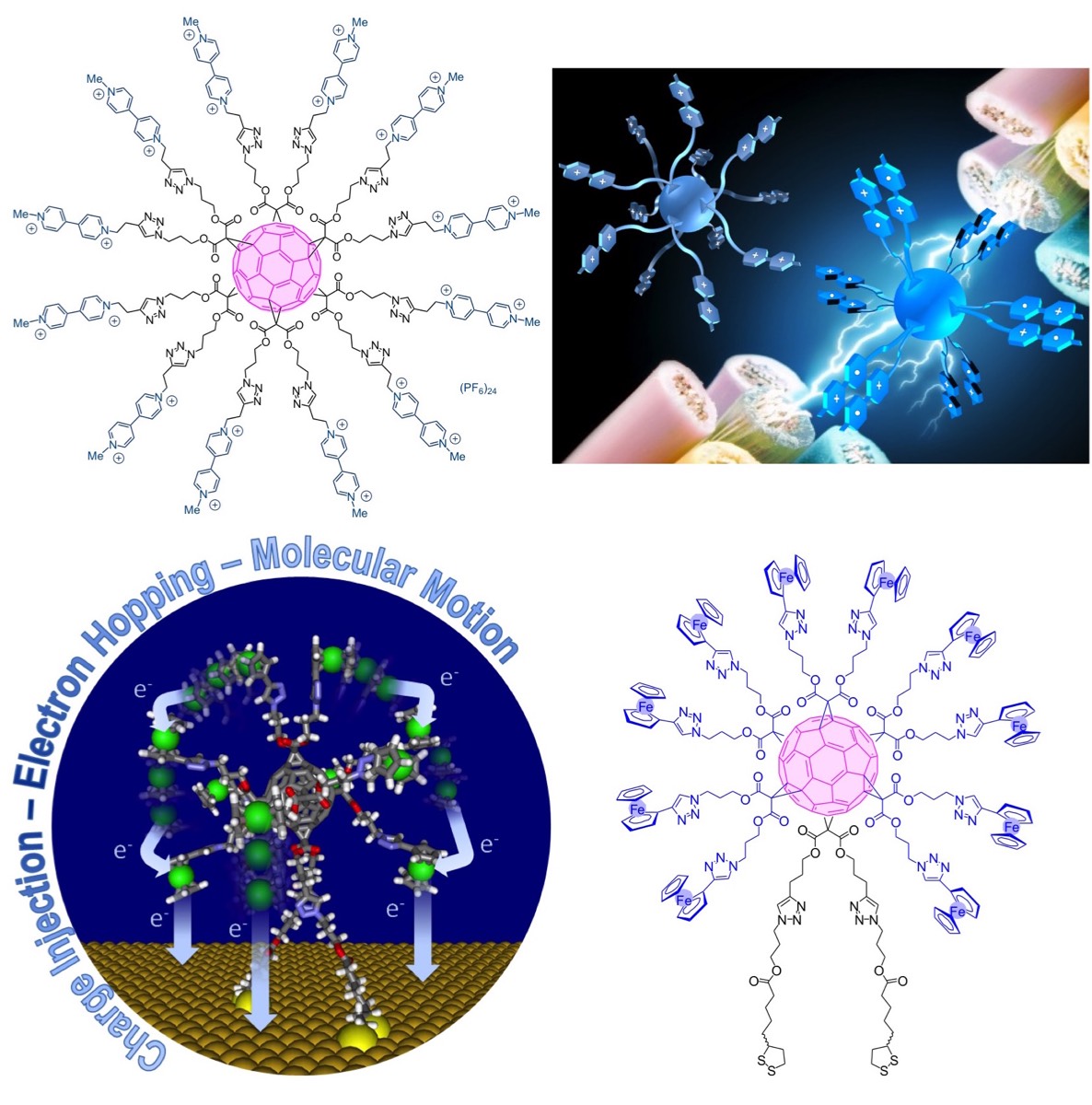
The characteristic electronic properties of [60]fullerene such as facile multiple reducibility or efficient singlet oxygen sensitization are essentially maintained for most of the fullerene derivatives. Even if fullerene mono- and bis-adducts present poorer electron acceptor properties than the parent [60]fullrene as a consequence of the saturation of one or two bonds of the fullerene framework that raises their LUMO energy, these fullerene derivatives are still easily reduced. In contrast, this is not the case anymore for fullerene hexa-adducts with a Th-symmetrical addition pattern. When compared to [60]fullerene, the first reduction of hexa-substituted fullerenes is dramatically shifted towards more negative potentials (by ca. 1 V). As a result, the fullerene core of hexa-adducts is electrochemically silent over a large potential window (from ca. -1.5 to +1.5 V vs. SCE). The hexa-substituted fullerene framework is therefore an attractive scaffold for the design of systems bearing peripheral electroactive subunits in a confined environment.